
Por Dra. Louize Galletti, Oftalmologista
Hoje, quero conversar com vocês sobre um tema muito importante na oftalmologia e que frequentemente gera dúvidas e apreensão no consultório: a Retinose Pigmentar (RP).
Embora o termo “retinitis pigmentosa” sugira que estamos lidando com uma inflamação, é fundamental esclarecer logo de início que a RP não é uma doença primariamente inflamatória. Trata-se, na verdade, de um grupo de doenças genéticas, também conhecidas como distrofias de cones e bastonetes, que afetam a retina de forma progressiva. A prevalência global dessa condição é de aproximadamente 1 caso para cada 4000 pessoas.
Como oftalmologista, meu papel é traduzir a complexidade dessa condição para você, paciente ou familiar, mostrando que, com o acompanhamento correto e a tecnologia atual, há caminhos para preservar a função visual e manter a qualidade de vida.
Os Primeiros Sinais: A Cegueira Noturna e a Visão em Túnel
A Retinose Pigmentar costuma dar seus primeiros sinais ainda na primeira ou segunda década de vida. Os dois sintomas clássicos que observamos são:
- Nictalopia (Cegueira Noturna): É muito comum o paciente relatar desorientação em ambientes pouco iluminados e uma tendência a esbarrar em objetos à noite. Esse sintoma ocorre devido ao comprometimento inicial dos bastonetes, as células da retina responsáveis pela nossa visão no escuro.
- Perda de Campo Visual: Inicia-se de forma lenta e progressiva na periferia da visão, geralmente simétrica entre os dois olhos. Com a evolução inexorável da condição, o paciente desenvolve o que chamamos de “visão em túnel”.
Além disso, entre 35% e 93% dos pacientes relatam fotopsias (flashes de luz), que são percebidas como pontos cintilantes ou arcos, podendo até mesmo ser contínuas e aumentar com o estresse. A alteração na visão de cores também pode surgir, tornando-se frequente quando a acuidade visual diminui.
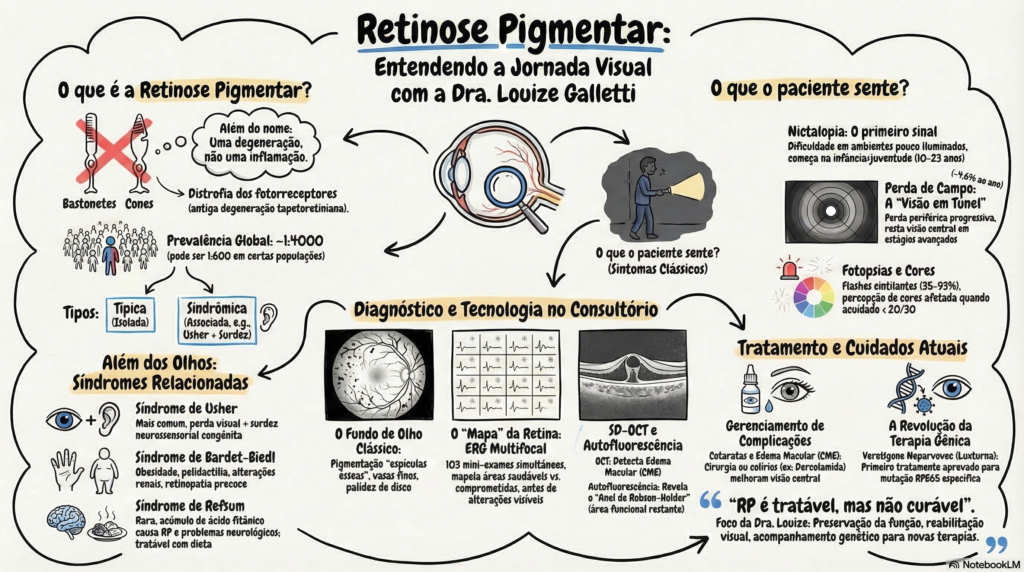
O Que Eu Vejo no Exame de Fundo de Olho?
Durante a consulta, o exame de fundo de olho é revelador. Os achados clássicos da RP incluem uma pigmentação que lembra espículas ósseas (resultado da migração de pigmento para a retina), vasos sanguíneos muito finos (atenuação vascular) e uma palidez no disco óptico.
Porém, em estágios iniciais, o fundo do olho pode parecer completamente normal, uma fase que chamamos de RP sine pigmento, que pode durar anos.
O Poder do Diagnóstico de Alta Tecnologia
Para entender exatamente como a doença está afetando a sua visão, utilizamos exames complementares de alta precisão:
- Tomografia de Coerência Óptica (SD-OCT): Esse exame de imagem moderno nos permite ver as camadas da retina em detalhes microscópicos. É a ferramenta de eleição para detectar a perda das camadas externas e identificar o Edema Macular Cistoide (CME), uma complicação comum na RP.
- Eletrorretinografia (ERG): Avalia a resposta elétrica da retina. Em especial, o ERG multifocal é um exame fascinante! O paciente olha para uma tela com 103 pequenos hexágonos que mudam de cor rapidamente. Em apenas 4 a 8 minutos, o computador cria um verdadeiro “mapa funcional” da retina, mostrando-me exatamente quais áreas estão saudáveis e quais estão comprometidas.
- Campimetria (Perimetria): Uma ferramenta essencial para quantificarmos a progressão da perda do campo visual ao longo dos anos.
A Genética por Trás da Retinose Pigmentar
Hoje, a forma mais moderna de classificar a RP é pelo gene mutado. Isso é vital porque nos permite prever a forma como a doença será herdada, o prognóstico e a possibilidade de terapias-alvo.
A maioria dos casos ocorre de forma isolada, mas a RP também pode ser “sindrômica”, ou seja, associada a condições sistêmicas. O exemplo mais comum é a Síndrome de Usher, que afeta cerca de 18% dos pacientes com RP e combina a doença ocular com surdez congênita.
O Tratamento: Preservando a Visão e a Qualidade de Vida
No consultório, costumo enfatizar uma ideia-chave: a Retinose Pigmentar é tratável, embora ainda não seja curável. Jamais aceite a frase “não há o que fazer”.
Nosso foco absoluto é preservar a sua função visual, tratar complicações e oferecer reabilitação:
- Manejo de Complicações: Pacientes com RP têm maior chance de desenvolver catarata (do tipo subcapsular posterior). A cirurgia de catarata é frequentemente indicada para melhorar a acuidade central. O Edema Macular Cistoide, que afeta cerca de 20% dos pacientes, é tratado incialmente com colírios específicos, como a dorzolamida, ou medicações via oral.
- Suplementação Vitamínica: Muito se fala sobre a Vitamina A. Estudos antigos mostraram uma modesta desaceleração na perda visual, mas o uso exige extrema cautela e monitoramento rigoroso devido aos riscos de toxicidade hepática e efeitos ósseos. A suplementação não é um padrão universal e deve ser discutida caso a caso.
- O Futuro Chegou (Terapia Gênica): Já existe uma terapia gênica aprovada chamada voretigene neparvovec (Luxturna™), destinada exclusivamente a pacientes com uma mutação específica (deficiência bialélica no gene RPE65), que tem mostrado melhora significativa na navegação em baixa luminosidade.
Um Alerta Importante: Devemos sempre desencorajar terapias não comprovadas cientificamente, como “curas milagrosas” ou tratamentos com células-tronco fora de estudos regulamentados, pois eles apresentam riscos de complicações graves e ausência de benefícios comprovados.
Se você ou alguém da sua família tem histórico de dificuldade visual noturna, perda de campo visual ou diagnóstico de Retinose Pigmentar, o acompanhamento oftalmológico regular e o aconselhamento genético são fundamentais.
Agende sua avaliação. Estamos juntos na missão de cuidar da sua visão com empatia, ciência e a melhor tecnologia disponível.