Por Dra. Louize Galletti
Olá! Sou a Dra. Louize Galletti e preparei este artigo com muito cuidado para desmistificar um tema complexo, mas de extrema importância na oftalmologia: a Retinose Pigmentar (RP) e suas doenças associadas. Se você ou alguém da sua família foi diagnosticado com essa condição, ou se você simplesmente deseja entender melhor como certas doenças genéticas afetam os olhos, este conteúdo foi feito para você. A informação é a nossa maior aliada para um acompanhamento oftalmológico de excelência.
O que é a Retinose Pigmentar?
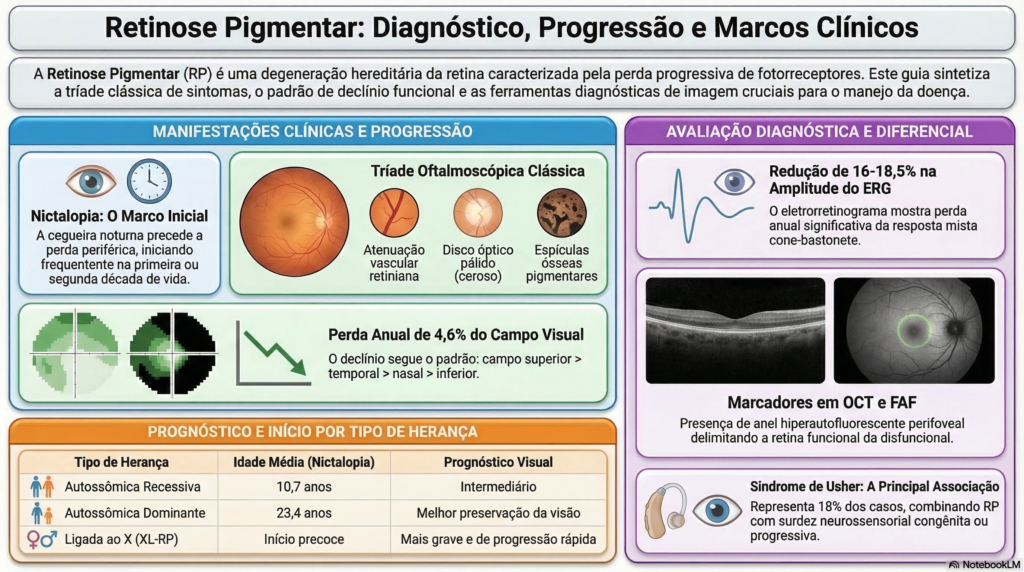
A Retinose Pigmentar é uma condição genética que afeta diretamente a retina. As duas marcas principais da RP são a dificuldade de enxergar à noite, condição que chamamos tecnicamente de nictalopia (cegueira noturna), e a perda progressiva e insidiosa do campo visual periférico.
Geralmente, essa dificuldade de visão noturna se inicia precocemente, na primeira ou segunda década de vida, o que infelizmente torna os pacientes mais propensos a acidentes. A perda do campo visual segue um padrão previsível ao longo dos anos: os déficits geralmente são mais graves no campo visual superior, avançando para a região temporal, nasal e, por fim, inferior. Na fase final, o padrão de visão torna-se “tubular”, como se o paciente estivesse enxergando através de um canudo, restrito aos 20 graus centrais ou menos.
Apesar de a periferia da visão ser o alvo principal, a visão central também pode ser seriamente afetada logo no início da doença devido a complicações como o edema macular cistoide (EMC), vazamento vascular retiniano e fibrose. Além disso, cerca de 35% dos pacientes percebem fotopsias, que são flashes de luz contínuos adjacentes às áreas de perda visual, e que tendem a desaparecer conforme a doença avança.
O Que Vejo no Consultório: Do Fundo de Olho aos Exames de Imagem
Durante a avaliação no consultório, buscamos achados clássicos no exame de fundo de olho: a atenuação (afinamento) dos vasos retinianos, palidez do disco óptico e o surgimento de pigmentações intrarretinianas com formato de “espículas ósseas”. No entanto, é importante destacar que a RP pode passar por um estágio chamado sine pigmento, onde a retina parece normal por décadas antes de as alterações típicas aparecerem, o que pode atrasar o diagnóstico, especialmente em pacientes que já possuem alta miopia. No segmento anterior do olho, é comum identificarmos o desenvolvimento de catarata subcapsular posterior em adultos jovens, entre 20 e 39 anos, além de alterações refracionais frequentes como alta miopia e astigmatismo.
A tecnologia é nossa grande parceira. O uso do SD-OCT (Tomografia de Coerência Óptica) demonstra com precisão a diminuição da espessura das camadas externas da retina, especialmente fora da mácula. Também utilizamos a autofluorescência (FAF), que revela a presença de um anel aumentado ao redor da fóvea, demarcando exatamente a fronteira entre a área de retina que ainda funciona e a área já disfuncional.
Para confirmar e classificar a doença, utilizamos exames funcionais. A perimetria (exame de campo visual) nos mostra escotomas relativos na média periferia que se aprofundam e formam um anel de perda visual. Já o Eletrorretinograma (ERG) é essencial para avaliar a atividade elétrica da retina, revelando, por exemplo, perdas difusas da função dos fotorreceptores (bastonetes) nas fases precoces.
Doenças e Síndromes Associadas
A Retinose Pigmentar muitas vezes não atua sozinha e pode estar acompanhada de alterações em outras partes do corpo. A associação sistêmica mais conhecida é a Síndrome de Usher, que afeta cerca de 18% dos pacientes com RP e se caracteriza pela combinação da retinopatia com surdez congênita ou progressiva. O diagnóstico precoce dessa síndrome é vital, pois o implante coclear pode ser muito bem-sucedido na preservação da audição e fala.
Outras condições sistêmicas relevantes incluem:
- Amaurose Congênita de Leber (LCA): Causa deficiência visual grave desde a infância, acompanhada frequentemente de nistagmo e um ERG profundamente anormal ou ausente. Associa-se comumente ao ceratocone e ao hábito de esfregar os olhos fortemente (sinal oculodigital).
- Síndrome de Bardet-Biedl: Uma doença autossômica recessiva que cursa com retinopatia, polidactilia (dedos a mais), obesidade congênita e, de forma muito comum, anormalidades renais. Ao contrário da RP clássica, a visão central é ruim desde o início.
- Doença de Refsum: Uma condição associada ao acúmulo de ácido fitânico no organismo, causando problemas neurológicos, surdez, catarata e uma retinopatia pigmentar. A restrição do consumo de ácido fitânico na dieta (presente em laticínios) pode limitar a progressão da doença sistêmica.
- Distrofia de Cones e Bastonetes: Ao contrário da RP típica, aqui a perda inicial ocorre na visão central e na visão de cores, progredindo posteriormente para o campo periférico, e frequentemente apresentando uma lesão macular em formato de “olho de boi” (bull’s eye).
O Perigo das “Falsas Retinoses” (Pseudoretinites)
No papel de oftalmologista, meu dever é realizar um diagnóstico preciso, pois diversas outras doenças podem simular o aspecto da Retinose Pigmentar no fundo de olho, condição que chamamos de Pseudoretinitis Pigmentosa.
Infecções antigas como sífilis, rubéola congênita (que também cursa com surdez) e toxoplasmose podem gerar cicatrizes que lembram a RP, porém o ERG costuma ser muito menos afetado nestes casos infecciosos. A toxicidade por medicamentos de uso contínuo, como a tioridazina, cloroquina e a quinina, pode causar uma retinopatia severa.
Ainda mais grave são as Retinopatias Paraneoplásicas Autoimunes (CAR), onde o paciente desenvolve degeneração grave da retina secundária a um câncer em outra parte do corpo, frequentemente pulmão ou colo do útero, podendo a perda visual preceder o próprio diagnóstico do tumor. Por fim, existe a Neurorretinite Subaguda Unilateral Difusa (DUSN), causada pela infecção da retina por um verme, que na fase avançada gera acúmulo de pigmentos parecidos com os da RP.
O acompanhamento oftalmológico regular, munido de exames de ponta e conhecimento atualizado, é o que garante o direcionamento genético, o tratamento de complicações como cataratas e edemas maculares, e a máxima preservação da qualidade de vida de nossos pacientes.